

¿Qué son los descriptores moleculares?
Para poder generar modelos QSAR, es necesario “codificar” de algún modo las características de las moléculas, de manera que tengamos una descripción lo más detallada posible de las mismas. Dicho de otro modo, es necesario traducir la estructura de las moléculas a valores numéricos que puedan ser fácilmente entendidos por los algoritmos informáticos, de forma que éstos puedan seleccionar aquellas características más relevantes en la bioactividad estudiada. Estas características son lo que denominamos “descriptores moleculares”.
Pero, ¿cuáles son estos descriptores moleculares? Se han descrito hasta ahora multitud de ellos, algunos de una gran complejidad, en esta entrada vamos a intentar mencionar los más simples y fáciles de entender.
Pongamos de ejemplo la siguiente molécula:

De un solo vistazo, podemos definir varios descriptores basados en la composición de átomos, o bien algunos que tengan en cuenta grupos funcionales:

Tambén podríamos recopilar o calcular ciertas propiedades físicas, como puede ser el peso molecular o la solubilidad, para utilizarlas como descriptores.
Otras características moleculares no son tan intuitivas. Un ejemplo son los caminos moleculares o WalkCounts, que definen todos los caminos de una longitud dada que podemos hacer dentro de una molécula. Así, si volvemos a nuestra molécula ejemplo, podemos definir dos caminos de 4 átomos empezando desde el carbono situado en el extremo izquierdo (marcado con un asterisco), que vienen representados por la siguiente figura:

O tres caminos de 4 átomos si empezamos desde el carbono situado en el extremo superior, también marcado con un asterisco en la siguiente figura:

Otros descriptores se refieren a la conectividad de los átomos. Siguiendo con nuestro ejemplo y numerando los átomos, para que sea más fácil de calcular:
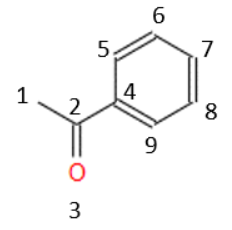
A partir de esta numeración podemos definir varios descriptores que hablan de la conectividad, por ejemplo, el número de átomos que están unidos a 3 átomos más, que, en este caso, sería 2 (los átomos 2 y 4). También podríamos calcular la conectividad global de la molécula o el número de oxígenos conectados con dobles enlaces. Y así hasta un montón de descriptores que tienen en cuenta este tipo de características. Una característica fundamental de todos ellos es que su valor es independiente de cómo se dibujen las moléculas, o por dónde se inicie la numeración de los átomos.
Hay otros muchos descriptores que no son tan intuitivos y fáciles de calcular a partir de la estructura dibujada. Por ejemplo, podemos utilizar como descriptor la energía del mayor orbital ocupado y la del menor orbital no ocupado. Es lo que se conoce como la energía HOMO (del inglés Highest Occupied Molecular Orbital) y la energía LUMO (del inglés Lowest Unoccupied Molecular Orbital).
A partir de una molécula pueden definirse multitud de descriptores. En la actualidad, nuestro software calcula cerca de 5.000 descriptores para cada molécula. De esta forma, antes de desarrollar un modelo QSAR podemos disponer de una gran matriz con una caracterización muy completa de cada estructura. Esta representación es unívoca: dos moléculas diferentes no pueden tener todos los descriptores iguales, de forma que cada estructura tiene su propia “huella digital” numérica.
Referencias
[1] Peukert, S., Nunez, J., He F. et al. A method for estimating the risk of drug-induced phototoxicity and its application to smoothened inhibitors. Med. Chem. Commun., 2011,2, 973-976. http://dx.doi.org/10.1039/C1MD00144B
[2] Gozalbes R., Doucet J. P. and Derouin F. Application of Topological Descriptors in QSAR and Drug Design: History and New Trends. Current Drug Targets – Infectious Disorders, 2002, 2, 93-102 93 . https://doi.org/10.2174/1568005024605909
[3] Todeschini R. and Consonni V. Molecular Descriptors for Chemoinformatics, Wiley- VCH, 2009. Online ISBN:9783527628766. https://doi.org/10.1002/9783527628766


