

What are molecular descriptors?
In order to generate QSAR models, it is necessary to codify in some way the characteristics of the molecules, in order to have the most detailed possible description of these. In other words, it is necessary to translate the molecular structures into numerical values that can be easily understood by computer algorithms, so that they the most relevant characteristics for the studied bioactivity are selected. These characteristics are what we call ‘molecular descriptors’.
But what are these molecular descriptors? Several descriptors have been proposed to date, some with a great deal of complexity, in this post we will attempt to mention the simplest and easiest to understand.
Let’s take the following molecule as an example:

At a glance, we can define several descriptors based on the composition of atoms, or some that take into account functional groups:

We could also compile or calculate certain physical properties, such as molecular weight or solubility, to be used as descriptors.
Other molecular characteristics are not so intuitive. An example is the molecular paths or Walk Counts, which define all the paths of a given length that may be identified for a given molecule. If we go back to our example molecule, we may define two paths of 4 atoms starting from the carbon located at the extreme left (marked with an asterisk), which are represented by the following figure:

Or three paths of 4 atoms if we start from the carbon located in the upper end, also marked with an asterisk in the following figure:

Other descriptors refer to the connectivity of atoms. Continuing with our example and numbering the atoms, to make it easier to calculate:
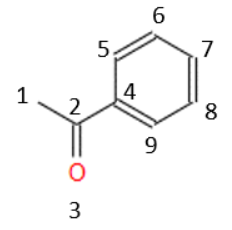
From this numbering we can define several descriptors that provide information on the connectivity, for example, the number of atoms that are attached to 3 or more atoms, which, in this case, would be 2 (atoms 2 and 4). We could also calculate the global connectivity of the molecule, or the number of oxygens connected by double bonds. In this way, there are a lot of descriptors that consider these types of characteristics. A fundamental characteristic of all of them is that their value is independent of how the molecules are drawn, or where the numbering of the atoms begins.
There are many other descriptors that are not so intuitive and easy to calculate from the drawn structure. For example, we can use the energy of the highest occupied and lowest unoccupied molecular orbitals as descriptors. These descriptors are known as the HOMO (Highest Occupied Molecular Orbital) and the LUMO (Lowest Unoccupied Molecular Orbital) energies. These descriptors are particularly important for certain toxicological properties, such as phototoxicity.
A multitude of descriptors may be defined from a molecule. Currently, our software calculates about 5,000 descriptors for each molecule. In this way, before developing a QSAR model we can have a large matrix with a very complete characterization of each structure. This representation is unique: two different molecules cannot have all the same descriptors, so each structure has its own numerical “fingerprint”.
References
[1] Peukert, S., Nunez, J., He F. et al. A method for estimating the risk of drug-induced phototoxicity and its application to smoothened inhibitors. Med. Chem. Commun., 2011,2, 973-976. http://dx.doi.org/10.1039/C1MD00144B
[2] Gozalbes R., Doucet J. P. and Derouin F. Application of Topological Descriptors in QSAR and Drug Design: History and New Trends. Current Drug Targets – Infectious Disorders, 2002, 2, 93-102 93 . https://doi.org/10.2174/1568005024605909
[3] Todeschini R. and Consonni V. Molecular Descriptors for Chemoinformatics, Wiley- VCH, 2009. Online ISBN:9783527628766. https://doi.org/10.1002/9783527628766


